
DP还能干这个?深度剖析DP在电池材料中的应用

研究背景介绍

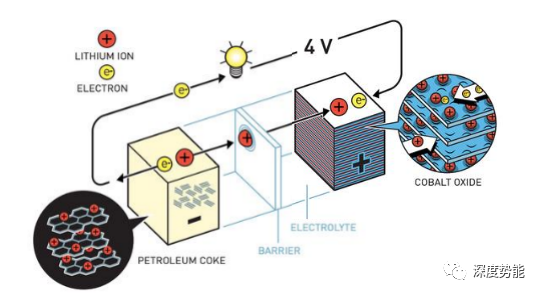
锂离子在正极-电解质-负极间往复运动。今天的商用锂离子电池主要由正极、电解液、隔膜和负极组成。有机电解液材料的易燃性是造成电池热失控的根本原因。使用不易燃的无机固体电解质的全固态电池体系,可以从根本上解决电池体系的安全问题,同时实现较高的能量密度。于此同时,要保证“充电一分钟通话两小时”,就需要固体电解质材料具有较高的离子电导率和扩散系数。以LiGePS类无机固体电解质为代表的硫化物材料是目前公认的下一代电池的主要技术路线。要优化并设计新型的固体电解质材料,研究者们需要从根本上理解锂离子在电池材料中的扩散行为,尤其是存在协同扩散行为的固体电解质材料。固体核磁,中子衍射等实验手段可以从原子尺度揭示锂离子在不同位点间扩散的方式,然而,由于成本和研究效率限制,实验表征手段不适宜于高通量的材料设计,例如材料组分精确调控。与之相对,低成本的原子尺度的计算模拟可以提供微观尺度的物理图像,为研究者们理解材料反应,设计优化电池材料提供了理性依据。

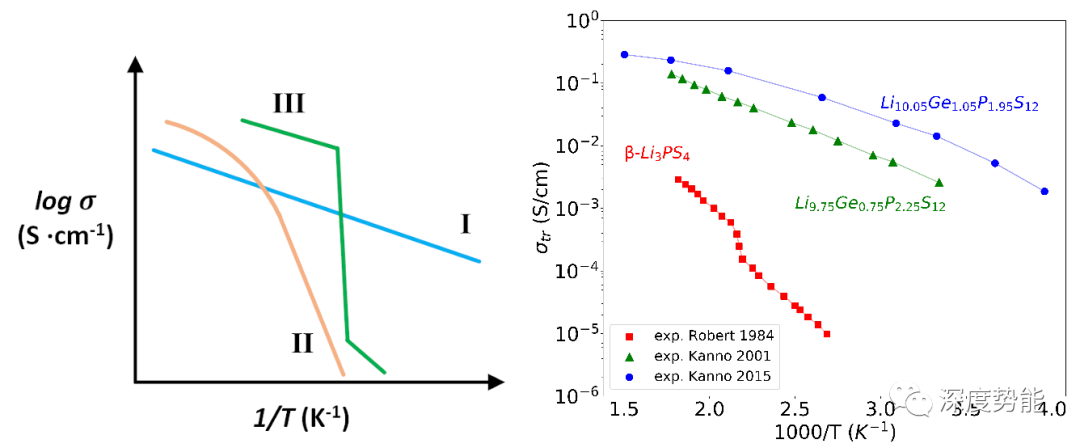
目前,对电池材料的扩散行为的模拟主要基于两类方法:基于NEB方法的热力学计算和基于第一性原子分子动力学(AIMD)的扩散过程模拟。NEB方法通过分析可能的扩散路径上的静态构象,模拟单原子或两原子运动的势能面以推算扩散势垒(Ea)。其计算成本低,适用于分析单原子在低浓度下的扩散行为,但是不适宜于模拟较高离子浓度条件下的协同扩散行为。基于AIMD的方法可以直接模拟扩散行为,对固体电解质材料中普遍存在的协同扩散行为进行模拟和解释,然而,由于AIMD计算成本高,目前使用AIMD计算模拟的体系大小一般不超过200个原子,模拟时长在100-200 ps左右,并且无法直接模拟室温下的扩散行为,只能通过在高温下模拟扩散过程再线性外推预测室温下的扩散性质。以固体电解质材料为例,研究已经发现了三类在高温和低温具有不同扩散行为的材料体系。高温外推法只适用于不发生相变或准相变行为的体系。以实验手段相比照而言,目前常用的计算模拟方法是“非原位的”,不能实现对实验条件下电池材料的准确模拟。 简而言之,算的准的算不快(AIMD),算的快的算不准(NEB)。如何才能二者兼具呢?近年来快速发展的机器学习势函数方法为又快又准地模拟扩散行为提供了可能。

机器学习势函数的自动化训练与评估
机器学习势函数方法通过满足一定几何和物理限制的变换,将每个原子的局域结构(local environment)表示为一个描述符(descriptor),再应用机器学习方法(如高斯过程,神经网络等)拟合其与高精度的第一性原理数据(能量,力等)间的关系,从而获得自适应的势函数(力场)。通过不断的从第一性原理数据中学习迭代提高势函数的精度和泛用性。当势函数迭代收敛后,可以应用于大体系长时间的准确模拟。当前适用于材料体系模拟的机器学习势函数方法主要有Belher-Parrinello Neural Network(BPNN), Gaussian Approximation Potential(GAP), aenet, Deep Potential(DP)等。机器学习势函数方法已被成功应用于材料物理化学性质的模拟,实现千万原子体系纳秒尺度以上快速模拟,极大的拓展了原子尺度高精度模拟的边界。
构造高精度的力场以往需要专业的研究人员进行长期开发优化,目前广泛使用的模拟软件如LAMMPS/Gromacs等等自建的力场都来源于专业人士的长期开发和积累。机器学习势函数方法,使得力场构建这一“旧时王谢堂前燕”,现在也能“飞入寻常百姓家”。尽管如此,在实践中,要快速构造材料的势函数并实现高精度模拟,还需要关注:
- 自动化的计算和数据处理流程。为了实现势函数的自动化构造,计算的所有步骤应当能自动化。
- 由于材料体系往往含有多种元素,基于原子环境的机器学习势函数应当能处理多元素体系且计算成本可控。由于不同机器学习势函数构造方法和代码实现的区别,在计算多元素体系时,计算性能往往有数量级的差异。
- 应用于实际模拟问题时,需要针对所研究问题建立一套高效、稳定的采样手段。保证势函数训练的稳定性。

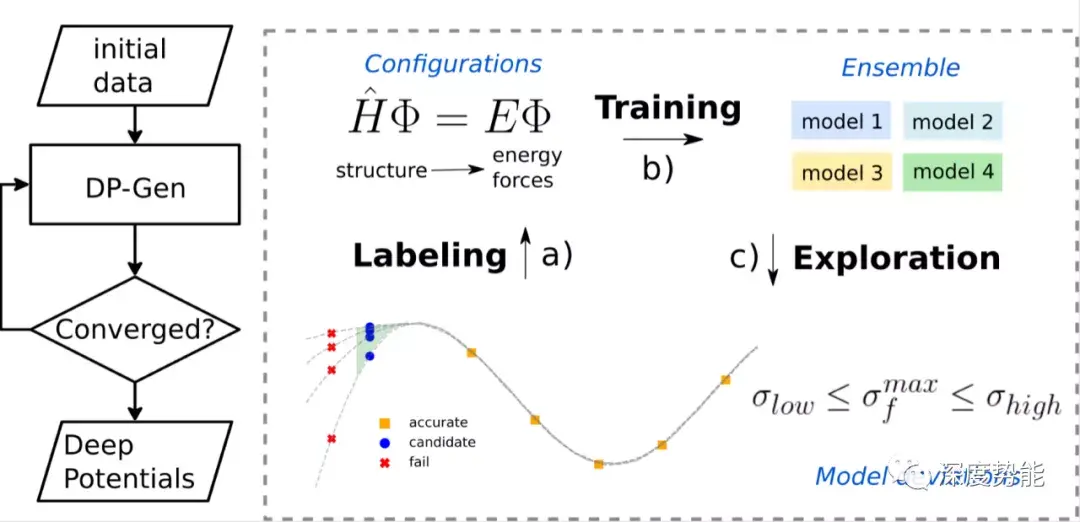
得益于近年来计算材料学领域的迅猛发展,尤其是材料基因组项目的建设,推动了高效且标准化的开源材料数据处理工具的开发,使得材料计算模拟更加自动化。这些工具为机器学习势函数相关的代码进一步开发提供了坚实基础。目前,在机器学习势函数,基于Deep Potential 的Deep Potential GENerator (DP-GEN)软件包提供了自动训练势函数的开源解决方案。由于其基于End-to-End的神经网络架构,计算复杂度随元素种类数目接近线性增长,快于一般的机器学习势函数方法。因此,我们在研究中选用DP-Gen来构建无机固体电解质体系的势函数并模拟确立可靠的锂离子扩散过程研究protocol。
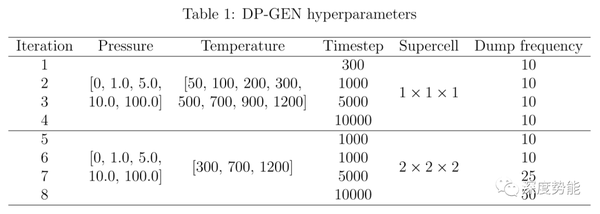
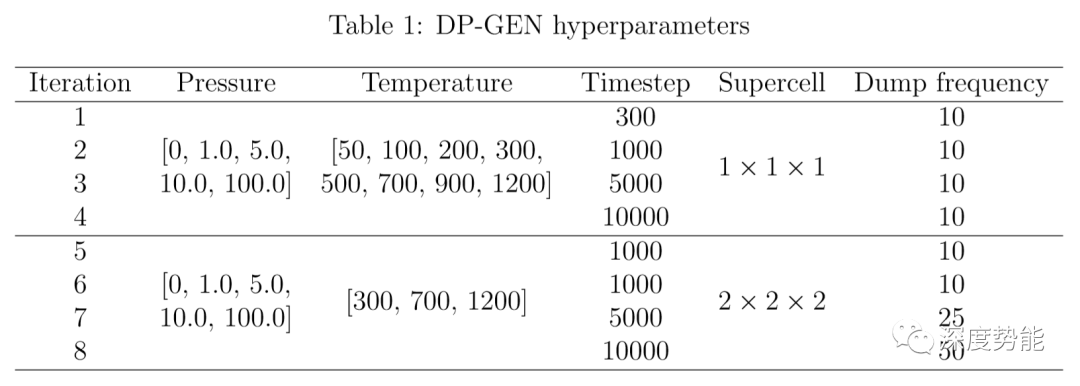
针对具有较高离子电导率的硫化物系固体电解质(Li10GeP2S12,Li10SiP2S12,Li10SnP2S12),通过同时进行多组不同温度的分子动力学模拟(MD)并扩展超胞(supercell)进行探索采样,预设8轮迭代训练。在DP-GEN迭代过程中,一般同时训练4个具有不同随机种子的势函数,根据4个势函数模型所预测的新构象的能量或力的方差选择新样本。

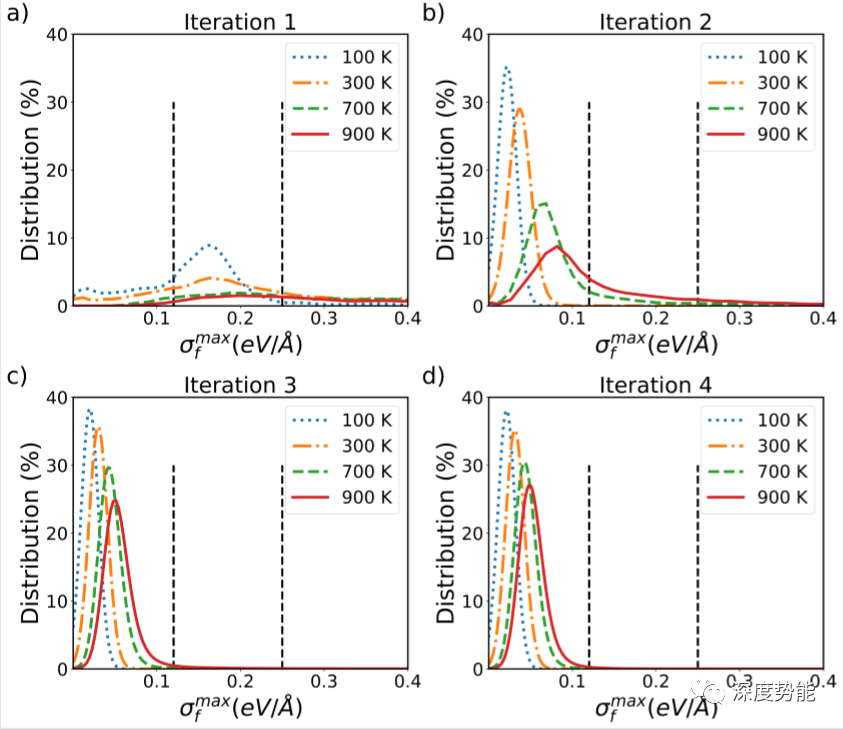
通过分析,可以直观的发现:仅仅通过三轮MD的探索,势函数的训练就已经快速收敛,新样本所占数目在5%以下。这是由于在固体电解质材料中,骨架原子主要在平衡位置附近振动,而锂离子具有较高的扩散系数,呈现类似液体的动力学性质。体系的自由度主要由锂离子决定。因此基于MD探索的DP-GEN可以快速采样对应的构象空间并训练势函数。

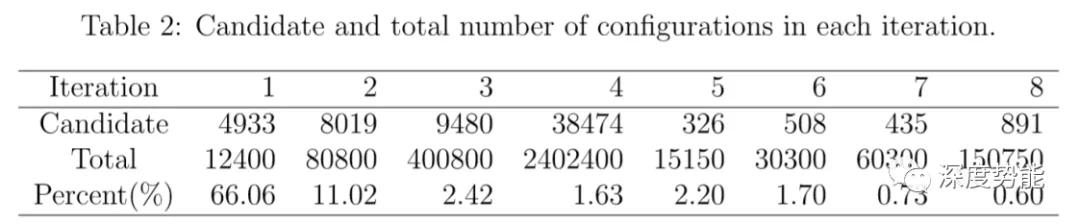
经过8轮的训练,在大体系中,所能找到的新构象比例已经很低,可以认为势函数已经收敛。获得势函数后,一般需要对比其和第一性原理计算数据的误差。在训练所得的第一性原理计算的数据集来评估势函数,其能量和力的误差大致在1meV/atom和80meV/A。可以认为势函数已经足够精确,可以应用于长时间大尺度的计算模拟。总共需要2500个左右的单点能计算。


使用Deep Potential模拟固体电解质材料
由于AIMD方法计算的尺度限制,计算所得到扩散系数有较大的统计误差。基于高温外推法所估计的扩散系数的误差可达10~100倍。为了获得统计可靠的扩散系数,需要进行充分的统计。在相关的研究中,主要需要考虑模拟的晶胞大小和模拟时长。为此,分别针对二者进行统计分析,查看收敛性。

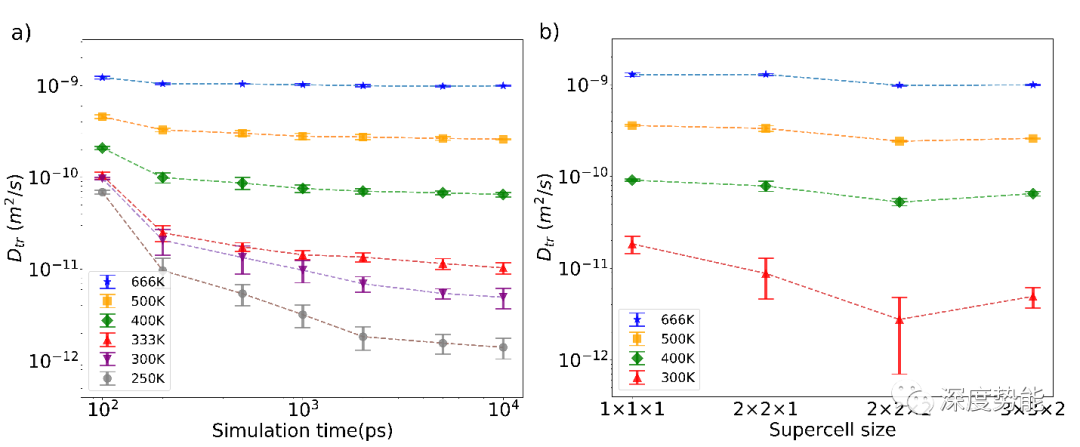
通过统计分析可以得出。至少需要900个原子的体系,10 ns以上长时间的分子动力学模拟才能可靠模拟室温下固体电解质材料内锂离子的扩散行为。在实际计算中,900原子体系10 ns的模拟使用了一张NVIDIA 2080TI 显卡,耗时70小时。

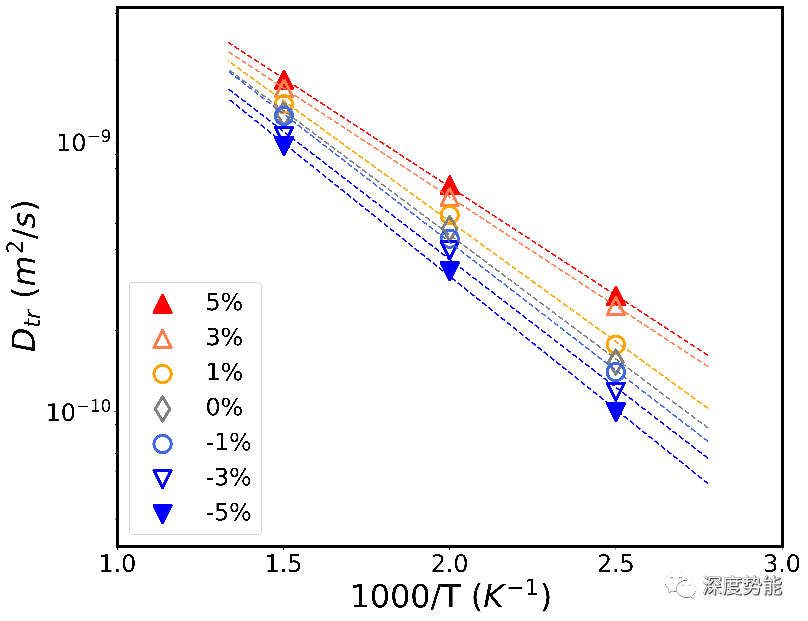
在无机固体电解质材料体相内,锂离子在四面体和八面体单元组成的孔道内定向扩散,扩散行为受到晶格常数调控。通过放缩晶胞体积,研究对比了不同晶格常数对扩散系数的影响。缩小晶胞会显著的降低扩散系数,而扩大晶胞带来的增益有限。因此,在实际模拟并对比固体电解质材料扩散系数时,应注意晶胞常数的选择。在实际的计算中,我们选用实验所测的晶格常数进行扩散过程模拟。

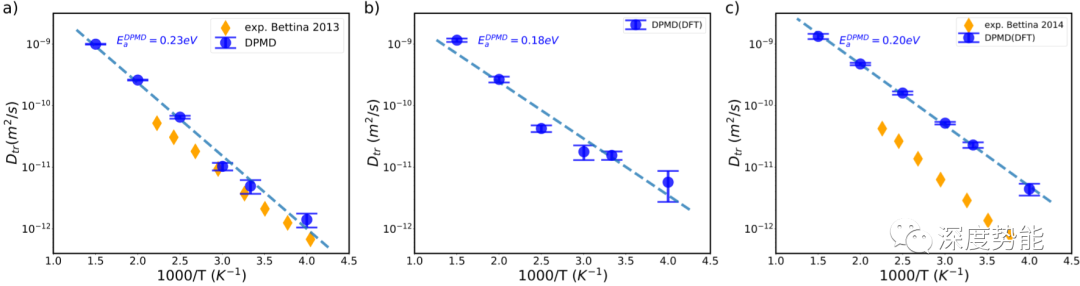
在明确了以上条件之后,我们模拟了三种固体电解质材料在250K-1000K范围内的扩散系数。计算所得扩散能垒与实验值较为吻合。计算所得扩散系数略高于实验所测扩散系数。这可能是因为实验中硫化物固体电解质材料的无序占据(Ge和P位点分数占据)和晶界电阻的影响,以及计算中所选泛函等因素影响。进一步的准确模拟需要考虑这些因素并对计算参数进行更详细的benchmark。对于Deep Potential模拟所得到的扩散数据,可以结合诸如GdyNN等数据分析方法进行分析,对扩散模式和扩散路径进行定量分析,以获得对扩散机制的详细理解。
展望
天下武功,唯快不破。基于原子局域结构表示的机器学习势函数方法极大的加速了材料模拟,推动原子尺度高精度模拟向纳秒和纳米以上发展,为多尺度材料模拟提供了一个可能的解决方案。以DP-GEN为代表的一类势函数工具,极大的便利了研究者开展相关的工作。
在目前探索的基础上,自然而然会提出这样的问题:在保持第一性原理计算精度的条件下,机器学习势函数所能达到的尺度边界在哪里?我们可以研究多大的体系,可以将对应的模拟时间延伸到什么尺度?在解答了这些问题之后,我们可以研究哪些重要的科学问题?基于大数据方法的新计算模拟工具为材料模拟带来了新的机遇,我们期待在后续的相关研究中结合机器学习方法和物理化学模型,解决电池材料领域的重要科学和技术问题。
原标题《固体电解质材料机器学习势函数的自动构建和扩散过程研究》,作者黄剑兴
原文:2020-08-19,审校:2021-01-30
