

化学热力学基础

一、化学热力学的研究范围、特点、局限性与意义
(1)研究范围
化学热力学研究化学变化的方向和限度,以及其伴随的变化过程中能量的相互转化所遵循的规律。化学热力学是一门宏观学科,研究一些热力学状态函数,不涉及物质的微观结构。
(2)特点
| 化学热力学解决化学反应过程中能量是如何转化的——能量守恒定律(热力学第一定律) |
| 化学热力学解决在指定条件下,化学反应朝哪个方向进行,以及反应进行的限度(热力学第二定律) |
(3)局限性
对于研究个别分子、原子的性质,即微观粒子的性质无能为力,所以对于研究对象无需知道其微观进程,也不需要考虑结构。
化学热力学研究问题时也没有时间概念,不涉及变化过程中速率问题。
(4)研究意义
了解反应的热效应在化工生产中非常重要,关系到如何开发能源和合理地利用能源。
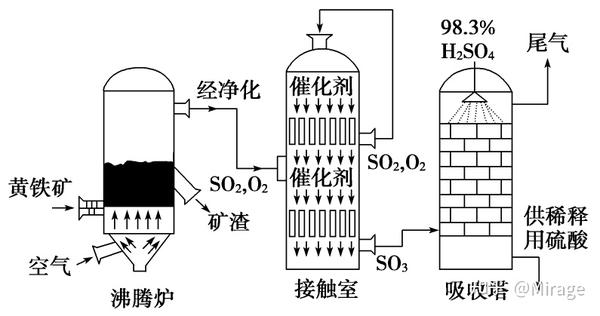
例如“接触法制硫酸”时, SO_2 和 O_2 在接触催化剂前必须预热,而 SO_2 氧化成 SO_3 又是放热反应,温度太高不利于 SO_3 的生成。
所以,就需要合理设计生产流程,利用热交换器把 SO_2 氧化反应产生的热量传递给需要预热的 SO_2 和 O_2 的混合气,以充分利用反应热,降低生产能耗。
二、热力学的基本术语
(1)体系与环境
热力学把被划定的研究对象称为体系,而与体系密切相关、有相互作用或影响所能及的部分称为环境。
H_2 与 Cl_2 反应生成 HCl ,体系为 H_2 、 Cl_2 、 HCl ,而环境为反应容器、周围空气等。
根据体系与环境之间的关系,把体系分为三类:
| 敞开体系(开放体系):体系与环境之间既有物质交换,也有能量交换。经典热力学不研究敞开体系。 |
| 封闭体系:体系与环境之间无物质交换,有能量交换。经典热力学主要研究封闭体系。 |
| 孤立体系:体系与环境之间无物质交换,无能量交换。实际上孤立体系不存在,只是一种理想模型。 |
以 Zn 和盐酸反应为例: 1° 反应在敞口容器中进行,则是敞开体系。 2° 反应在密闭容器中进行,则是封闭体系。 3° 反应在绝热的密闭容器中进行,并将容器置于真空中,则可看作是孤立体系。
(2)状态,状态函数
|
状态:由一系列表征体系性质的物理量总和所确定下来的体系的一种存在方式,称为体系的状态。
① 当体系的一系列宏观性质都为定值,不随时间变化时,体系就处于一定的状态;② 当其中一个物理量改变时,状态就发生改变。 |
| 状态函数:确定体系状态的物理量是状态函数,主要有内能、焓、熵、吉布斯自由能等。 |
状态函数的性质:其变化只取决于始态和终态,与途径无关;当系统恢复原态,状态函数一定恢复原值。
可以概括为: 异途同归,值变相等,周而复始,数值还原
* 广度量与强度量
描述均匀物体性质的物理量有两类:
| 广度量(广延量):与物质的总量成正比的:体积,内能,熵等等。 |
| 强度量:反映物质内在性质而与总质量无关的:压强,温度,比热等。 |
(3)过程和途径
| 过程:体系的状态发生改变,变化前称为始态,变化达到的状态称为终态。从始态到终态,经历了一个热力学过程,简称过程。常见过程有:等温过程、等压过程、等容过程、绝热过程等。 |
| 途径:实现过程的方式称为途径。完成一个热力学过程,可以采取不同的方式。我们把每一个具体的方式称为一种途径。过程着重于始态和终态,而途径着重于具体方式。 |
常见过程有:
| ① 等温过程:ΔT=0 |
| ② 等压过程:Δp=0 |
| ③ 等容过程:ΔV=0 |
| ④ 绝热过程:Q=0 |
例如:某理想气体经过一个恒温过程,可以用多种不同的途径。

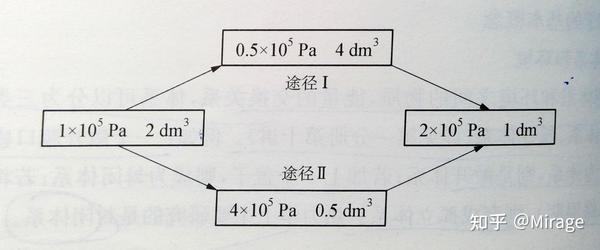
上述过程的两种途径中,
\begin{align*} &\Delta p=p_{final}-p_{primary}=2\times 10^5Pa-1\times 10^5Pa=1\times 10^5Pa \end{align*}
\Delta V=V_{final}-V_{primary}=1dm^3-2dm^3=-1dm^3
(4)功和热
在体系与环境发生能量交换时,能量有两种形式——热和功,均具有能量单位 J 或 kJ 。
① 体积功 W_e 与非体积功 W_f
化学反应过程中,经常发生体积变化。体系抵抗外压改变体积,产生体积功。
设在一截面面积为 S 的圆柱形筒内发生化学反应,体系反抗外压 p 膨胀,活塞从 I 位置移动到 II 位置。
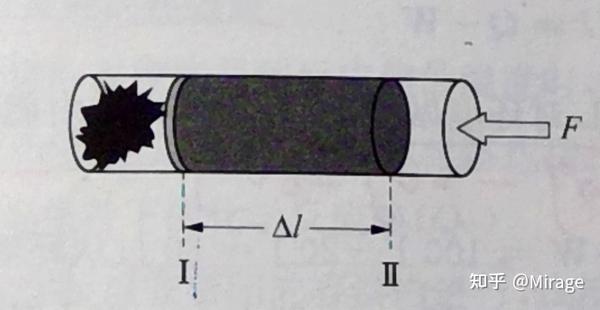
则,对于气体体系的体积功(膨胀功): W_e=-F\Delta l = -\frac{F}{S}·S\Delta l = -p\Delta V
规定:当环境对体系做功,功为正;当体系对环境做功,功为负。
这里研究的体系与过程,若不加特殊说明,可以认为只做体积功。
除体积功以外的所有其他形式的功统称为非体积功,如机械功( W=\vec{F}·\vec{s} )、电功( W=E_{电动势}q )等。
② 热
由于温度不同而在体系和环境之间传递的能量称为热,用 Q 表示。
规定:当热由环境流入体系, Q>0 ;当热由体系流入环境, Q<0
我们不难发现, 功和热符号的正负是从体系自身出发的 ,使体系热力学能增加的为正值,降低的为负值; 功和热不是状态函数 ,它们不能表征一个系统的状态,而用于描述能量转化的多少。
(5)热力学能(内能)
指系统内部能量的总和,包括分子运动的平动能、分子内的转动能、振动能、电子能、核能以及各种粒子之前的相互作用位能等。
热力学能是状态函数,用 U 表示,其绝对值无法测定,但我们只关心其变化量: \Delta U=U_{final}-U_{primary}
对于理想气体,不考虑分子间引力、分子距离,理想气体的内能只是温度的函数,即 U_{id}=f(T) 。
由“气体分子运动论”,我们知道: pV=\frac{m}{3}N\bar{v}^2=\frac{2N}{3}·\frac{1}{2}m\bar{v}^2=\frac{2N}{3}\bar{E_k}
又由于“理想气体状态方程”: pV=nRT ,得到 \Delta U=\Delta (pV)=\Delta (nRT)
所以,对于物质的量一定的理想气体: \Delta (pV)=\Delta U=nR\Delta T
三、热力学第一定律、热力学第二定律
(1)热力学第一定律(能量守恒定律) :
文字描述:自然界的一切物质都具有能量,能量不可能被创生,也不可能被消灭;能量有各种不同的形式,能够从一种形式转化为另一种形式,但在转化过程中,能量的总值保持不变。
数学表达式: \Delta U=Q+W
若系统发生微小的变化,热力学能的变化为: dU=\delta Q+\delta W
(2)* 热力学第二定律(Clausius 不等式) :
文字描述:热不可能自发地、无代价地从低温物体传至高温物体。
数学表达式: \Delta S_{A\rightarrow B}-\sum_A^B\frac{\delta Q}{T}\geq0
应用到微小过程: dS-\frac{\delta Q}{T}\geq 0
四、热化学
(1)化学反应的热效应( Q_v 与 Q_p )
当生成物的温度恢复到反应物的温度时,化学反应所放出或吸收的热量,称为化学反应热效应,简称反应热(无非体积功)
① 恒容反应热( Q_v )——不常见的反应热
若化学反应在密闭的容器中进行,该反应称为恒容反应。其热效应为恒容反应热 Q_v 。
恒容条件下反应,体系不会膨胀,于是不做体积功,体系的内能变化量等于体系吸收或放出的热量。
即 Q_v=\Delta_r U ( r 的下标代表 reaction ,反应)
即恒容条件下的反应热等于体系内能变化量。
若 \Delta_r U=Q_v>0 ,则称反应是吸热(体系能量升高);若 \Delta_r U=Q_v<0 ,则称反应是放热。
此处要注意和一般的 +Q,-Q 的不同,上面所说的反应热是对于体系而言,后者则对于环境而言。
② 恒压反应热( Q_p )——更常见的反应热
通常化学反应在敞口容器中进行,外界大气压一般为恒定的一个大气压。像这类在恒压过程中完成的化学反应,称为恒压反应。其热效应为恒压反应热 Q_p 。
恒压条件下, \Delta p = 0 ,体积功为 W_e=-p\Delta V
由热力学第一定律,有 \Delta_rU=Q_p+W_e=Q_p-p\Delta V=Q_p-\Delta (pV)
移项,则恒压反应热为: Q_p=\Delta_rU+\Delta (pV)=\Delta(U+pV)
(2)焓( H )
上面我们得到: Q_p=\Delta(U+pV) ,由于 Q,V,p 都是状态函数,所以 U+pV 也是一个状态函数。
不妨引入一个新的状态函数—— 焓 (热焓,enthalpy): H\xlongequal{def}U+pV ,于是 Q_p=\Delta_r H 。
归纳一下,现在我们知道了: \left\{\begin{array}{**lr**} Q_v=\Delta_r U\\ \\ Q_p=\Delta_r H=\Delta_r U+\Delta (pV) \end{array} \right.
下面讨论 Q_v,Q_p 的关系:
同一反应的 Q_v,Q_p 并不相同,所以如果确定了反应物和生成物,其反应热是不确定的,还需要给出条件。
对于理想气体而言,两个 \Delta _r U 是相等的;真实气体情况下,认为近似相等。
所以, Q_p=Q_v+\Delta (pV)=Q_v+\Delta (nRT)=Q_v+(\Delta n)RT ,此处反应前后温度是一致的。
这里的 \Delta n 为气体生成物的物质的量的总和与气体反应物的物质的量的总和之差。
那么, \left\{\begin{array}{**lr**} Q_p=Q_v+(\Delta n)RT\\ \\ \Delta_r H=\Delta_r U+(\Delta n)RT \end{array} \right.
对于无气体参与的固液体反应,一般 \Delta V 可以忽略,近似认为 Q_v=Q_p
(3)热化学方程式
① 热力学标准状态(标态):
选用 1\times10^5Pa \,\,(1\,\,bar) 作为气态物质的热力学标态,符号为 p^\Theta
溶液的标态则指溶质质量摩尔浓度或活度为 1\,\,mol·kg^{-1} 。
对于稀溶液,也可用物质的量浓度 1\,\,mol·dm^{-1} 。
② “1 mol 反应”:
把化学反应看成一个整体单元,发生“1 mol 反应”。
③ 标准摩尔反应焓: 参加反应的各物质都处于标准态时的反应焓变,记作 \Delta _r H_m^\Theta
如 {H_2}_{(g)}+\frac{1}{2}{O_2}_{(g)} \xlongequal[\quad]{}{H_2 O}_{(l)}\quad,\quad\Delta_r H_m^\Theta(298K) = -286kJ·mol^{-1}
或简写为 {H_2}_{(g)}+\frac{1}{2}{O_2}_{(g)} \xlongequal[\quad]{}{H_2 O}_{(l)}\quad,\quad\Delta H^\Theta = -286kJ·mol^{-1}
此处的 mol^{-1} 指的是 1\,\, mol 反应,即 1mol\quad({H_2}_{(g)}+\frac{1}{2}{O_2}_{(g)} \xlongequal[\quad]{}{H_2 O}_{(l)})
所以 2{H_2}_{(g)}+{O_2}_{(g)} \xlongequal[\quad]{}{2H_2 O}_{(l)}\quad,\quad\Delta_r H_m^\Theta(298K) = -572kJ·mol^{-1}
④ 标准摩尔生成焓: 在温度为T的标准态下,由指定单质生成1mol的某种物质,该生成反应的焓变即为该物质在温度T时的标准摩尔生成焓,记作 \Delta _f H_m^\Theta (f,formation,生成)
标准摩尔生成焓可用于计算反应焓变, \Delta_r H_m^\Theta=\sum_{生成物}n_i \Delta_fH_m^\Theta-\sum_{反应物}n_j \Delta_fH_m^\Theta
⑤ 标准摩尔燃烧焓: 在温度为T的标准态下,某物质与氧气进行完全氧化反应时,该生成反应的焓变即为该物质在温度T时的标准摩尔燃烧焓,记作 \Delta _c H_m^\Theta (c,combustion,燃烧)
标准摩尔燃烧焓可用于计算反应焓变,例如:
C_{_{石墨}}+{O_2}_{(g)} \xlongequal[\quad]{}{CO_2}_{(g)},\Delta H_1 = -394kJ·mol^{-1}\quad (1)
{H_2}_{(g)}+\frac{1}{2}{O_2}_{(g)} \xlongequal[\quad]{}{H_2 O}_{(l)},\Delta H_2 = -286kJ·mol^{-1}\quad (2)
C_2 H_5OH_{(l)}+3{O_2}_{(g)}\xlongequal[\quad]{}2{CO_2}_{(g)}+3{H_2 O}_{(l)},\Delta H_3 = -1367kJ·mol^{-1} \quad (3)
由 (1)\times2+(2)\times3-(3) 得:
2C_{_{石墨}}+3{H_2}_{(g)}+\frac{1}{2}{O_2}_{(g)}\xlongequal[\quad]{} C_2 H_5OH_{(l)},\Delta H=H_1\times2-H_2\times3-H_3=-279kJ·mol^{-1}
可见在过程中按照系数比约去了燃烧产物,并将生成物 C_2 H_5OH_{(l)} 移到了方程式右侧,得到了总反应方程式,总反应焓变即可表示。
于是归纳可得: \Delta_r H_m^\Theta=\sum_{反应物}n_i \Delta_cH_m^\Theta-\sum_{生成物}n_j \Delta_cH_m^\Theta
(4)盖斯定律
内容:一个化学反应,无论是一步完成,还是分多步完成,其热效应是相同的。
实际意义:有的反应虽然简单,但热效应难以测得。
例如: {C}_{(s)}+\frac{1}{2}{O_2}_{(g)} \xlongequal[\quad]{}{CO}_{(g)} 是很简单的反应,但很难保证反应产物的纯度。
于是考虑盖斯定律: \left\{\begin{array}{**lr**} {C}_{(石墨)}+{O_2}_{(g)} \xlongequal[\quad]{}{CO_2}_{(g)}\quad,\quad\Delta_r H_{m(1)}= -393.5kJ·mol^{-1}(1)\\ \\ {CO}_{(g)}+\frac{1}{2}{O_2}_{(g)} \xlongequal[\quad]{}{CO_2}_{(g)}\quad,\quad\Delta_r H_{m(2)}= -238.0kJ·mol^{-1}(2) \end{array} \right.
由 (1)-(2):{C}_{(石墨)}+\frac{1}{2}{O_2}_{(g)} \xlongequal[\quad]{}{CO}_{(g)}
\Delta_r H_{m}=\Delta_r H_{m(1)}-\Delta_r H_{m(2)}=-393.5kJ·mol^{-1}-(-238.0kJ·mol^{-1})=-155.5kJ·mol^{-1}
(5)熵( S )
熵是描述体系混乱程度的物理量,用符号S表示。物体的混乱程度越大,则熵值越大。
热力学第三定律: 在绝对零度时,任何理想晶体的熵值为0。以此为标准,可以计算任何物质在标准压力、指定温度时的熵值,即相当于该物质从 0 K 到 T K 的熵变 \Delta S^\Theta ,该熵变就是该物质在标准状态下的标准规定熵 S^\Theta 。
注意: 熵是状态函数。 物质的状态不同,熵值的差别较大。对于同一物质,气态的熵 > 液态的熵 > 固态的熵。 温度升高,物质的熵值越大;压力增大,物质的熵值减小(尤其对气体影响较大)。
对化学反应而言, \Delta_r S_m^\Theta=\sum_{生成物}n_i \Delta_fS_m^\Theta-\sum_{反应物}n_j \Delta_fS_m^\Theta
在孤立体系中,任何自发过程的结果都将导致体系的熵值增大。但由于孤立系统是一个理想体系,实际上难以实现,只有将体系和环境联合起来考虑,即 \Delta S_{孤立体系}=\Delta S_{体系}+\Delta S_{环境} ,那么用熵增加原理判断一个过程是否自发时,除了考虑体系的熵变以外,还要考虑环境的熵变,这样使用起来很不方便。
五、化学反应进行的方向
(1)自发过程与自发反应
自发过程: 在一定条件下不需要外力作用就能自动进行的过程。自然界中发生的过程都具有一定的方向性。如水从高往低处流;冰块室温下自动融化;铁器在潮湿空气中生锈;甲烷在空气中遇火燃烧。自发过程的逆过程是非自发的。
自发反应: 在给定条件下,一个反应可以自发地正向进行到显著程度,就称为自发反应。
注意: ① 自发过程(热力学可能)不一定迅速进行(动力学),如氢氧化合生成水。 ② 自发过程可以用来做功,如水力发电、甲烷燃烧用在内燃机中做功、设计原电池、氢燃烧设计成燃料电池等。 ③ 非自发反应并不意味着不能进行,环境对体系做功就可以使非自发反应进行,如水的电解。
(2)亥姆霍兹自由能
对于一个等温过程:
热力学第一定律: \delta Q=dU-\delta W
热力学第二定律: dS-\frac{\delta Q}{T}\geq 0
将热力学第一定律代入热力学第二定律: dS-\frac{dU-\delta W}{T}\geq 0
即 -\delta W\leq -(dU -TdS)
由于是等温过程, -\delta W\leq -d(U -TS)
定义亥姆霍兹自由能(功函): A\xlongequal{def}U-TS
由此可得: -\delta W\leq -dA 或 -W\leq -\Delta A
等温过程中,一个封闭系统所能做的最大功等于其亥姆霍兹自由能的减少。
系统在等温、等容且无其他功的情况下, -\Delta A\geq0 或 \Delta A\leq0
(3)吉布斯自由能
为了判断等温、等压化学反应的方向性,美国科学家吉布斯综合考虑焓和熵两个因素,提出一个新的状态:吉布斯自由能。
功分为两类,体积功和非体积功, -\delta W_e-\delta W_f\leq -d(U -TS)
pdV-\delta W_f\leq -d(U -TS)
-\delta W_f\leq -d(U +pV-TS)
由 H=U+pV ,得到: -\delta W_f\leq -d(H-TS)
定义吉布斯自由能: G\xlongequal{def}H-TS
由此可得: -\delta W_f\leq -dG 或 dG\leq \delta W_f
在等温、等压条件下,一个封闭体系所能做的最大非膨胀功等于其吉布斯自由能的减少。
对于化学反应,反应前后吉布斯自由能变化用 \Delta _rG_m 表示反应: \Delta _rG_m=\sum G_{产物}-\sum G_{反应物}
若 \Delta _rG_m<0 ,反应自发进行;若 \Delta _rG_m>0 ,则反应非自发;若 \Delta _rG_m=0 ,反应处于平衡态。
① 吉布斯方程及其应用
根据吉布斯自由能的定义式,在等温等压下可推导出吉布斯方程: \Delta G=\Delta H-T\Delta S
其把影响化学反应自发进行方向的两个因素 \Delta H,\Delta S 统一了起来。
不难看出 \Delta G 与 T 呈线性关系,若 \Delta H<0,\Delta S>0 ,则任意温度下, \Delta G <0 ,反应都能自发进行。
| △H | △S | △G | 反应方向 |
|---|---|---|---|
| - | + | <0 | 正反应恒定自发进行 |
| + | - | >0 | 正反应永不自发进行 |
| - | - | 低温时<0 | 正反应可以自发进行 |
| + | + | 高温时>0 | 正反应可以自发进行 |
② 标准摩尔生成吉布斯能
在标准状态下,由最稳定单质生成 1 mol 物质B的吉布斯自由能变,称为这种温度下B物质的标准摩尔生成吉布斯能,用符号 \Delta _fG_m^\Theta 表示。
由 \Delta _fG_m^\Theta 计算化学反应的 \Delta _rG_m^\Theta 的计算公式: \Delta _rG_m^\Theta=\sum_Bv_B\Delta _fG_m^\Theta(B)
温度对 \Delta _rG_m^\Theta 有影响,一定温度下化学反应的标准摩尔吉布斯能变化 \Delta _rG_{m,T}^\Theta 可按下式计算:
\Delta _rG_{m,T}^\Theta=\Delta _rH_{m,T}^\Theta-T\Delta _rS_{m,T}^\Theta
③ 非标准状态下的吉布斯自由能变的计算
非标准状态下化学反应的吉布斯自由能变,可由范特霍夫等温方程式求得:
\Delta _rG_{m,T}=\Delta _rG_{m,T}^\Theta+RT\rm lnQ=\Delta _rG_{m,T}^\Theta+2.303RT\rm lgQ
式中Q称为反应商。它是各生成物相对分压(对气体, \frac{p}{p^\Theta} )或相对浓度(对溶液, \frac{c}{c^\Theta} )幂的乘积与各反应物的相对分压或相对浓度幂的乘积之比。若反应中有纯固体或纯液体,则其浓度用常数1表示。
例如对于任意化学反应: aA_{(aq)}+bB_{(l)}\xlongequal{\,\,\quad} dD_{(g)}+eE_{(s)} , Q=\frac{(p_D/p^\Theta)\times 1}{(c_A/c^\Theta)\times 1}
在稀溶液中进行的反应,如果溶剂参加反应,因溶剂的量很大,浓度基本保持不变,可以当做常数1。
由表达式可知,Q的单位为1。
